{kind=link}
Read paper here
aKmerBroom is a tool to decontaminate ancient oral samples from a FASTA/FASTQ file. It does so in the following steps:
- Build an
ancient_kmers.bloomfilter from an ancient kmers text file (if such a Bloom filter does not yet exist). - For a set of input reads:
- Save those reads which have 2 consecutive kmer matches against
ancient_kmers.bloom - Kmerize the saved reads to generate a new set of ancient kmers, called "anchor kmers"
- Save those reads which have 2 consecutive kmer matches against
- For the same set of input reads, identify matches against anchor kmers and classify each read with >50% matches as an ancient read.
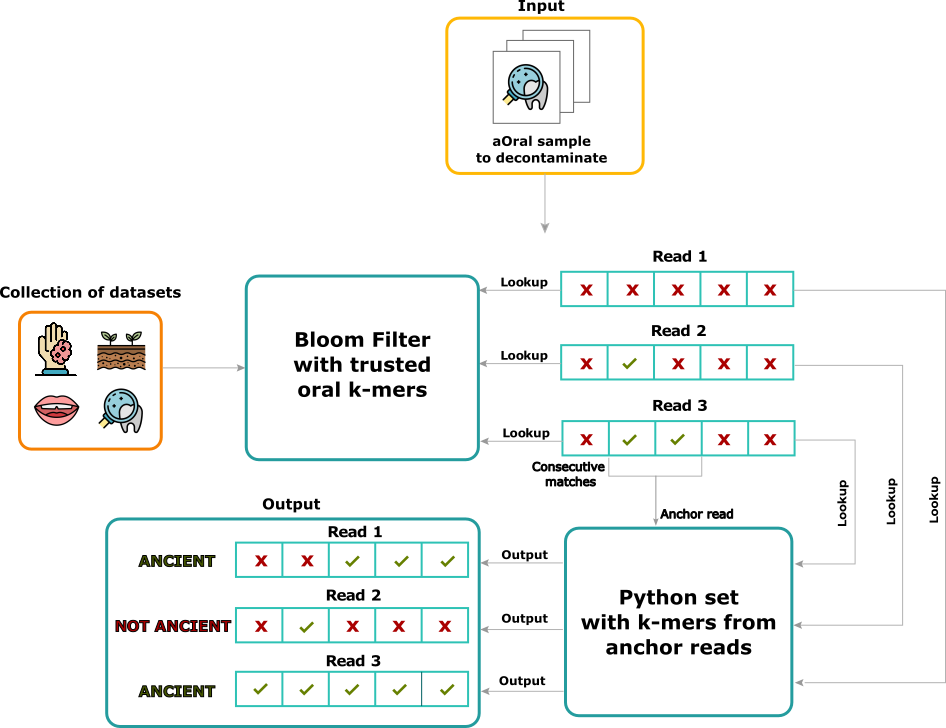 aKmerBroom pipeline: First, an offline step is performed: a collection of samples representative from diverse sources is used to create a trusted set of oral kmers. The trusted collection indexes kmers that appear exclusively in modern and ancient oral samples, but not other samples from contaminant sources (see panel on the left called Collection of datasets). Then this set of oral kmers is used to decontaminate an input set of reads. The algorithm proceeds by looking up each read kmer inside the Bloom Filter of trusted oral kmers, and marking positions of matches. Reads having at least two consecutive matches to the Bloom Filter get passed to the construction of a set containing all kmers from such reads. Finally, the same input reads are scanned again using the aforementioned set, and reads having a proportion of kmer matches over a certain threshold are reported to be of ancient oral origin.
aKmerBroom pipeline: First, an offline step is performed: a collection of samples representative from diverse sources is used to create a trusted set of oral kmers. The trusted collection indexes kmers that appear exclusively in modern and ancient oral samples, but not other samples from contaminant sources (see panel on the left called Collection of datasets). Then this set of oral kmers is used to decontaminate an input set of reads. The algorithm proceeds by looking up each read kmer inside the Bloom Filter of trusted oral kmers, and marking positions of matches. Reads having at least two consecutive matches to the Bloom Filter get passed to the construction of a set containing all kmers from such reads. Finally, the same input reads are scanned again using the aforementioned set, and reads having a proportion of kmer matches over a certain threshold are reported to be of ancient oral origin.
# Create the environment using mamba (faster than conda)
mamba env create -f environment.yml
# Activate the environment
mamba activate aKmerBroom
# Install aKmerBroom as a command-line tool
pip install -e .# Create and activate environment only
mamba env create -f environment.yml
mamba activate aKmerBroomAfter installation, you can run aKmerBroom --help to see all available options.
# Display help and see all arguments
aKmerBroom --help--input_file: Path to input FASTQ/FASTA file--output_prefix: Prefix for output files (enables batch processing)- One of:
--ancient_bloomOR--ancient_kmers_set
# Using pre-built Bloom filter
aKmerBroom --ancient_bloom --input_file /path/to/sample1.fastq --output_prefix sample1
# Using k-mers text file
aKmerBroom --ancient_kmers_set --input_file /path/to/sample1.fastq --output_prefix sample1# Custom k-mer size and thresholds
aKmerBroom --ancient_bloom --input_file sample.fastq --output_prefix sample1 \
--kmer_size 25 --anchor_proportion_cutoff 0.6 --output results/
# Batch processing
for file in *.fastq; do
prefix=$(basename "$file" .fastq)
aKmerBroom --ancient_bloom --input_file "$file" --output_prefix "$prefix"
done| Argument | Description | Default | Required |
|---|---|---|---|
--ancient_bloom |
Use pre-built ancient kmers Bloom filter | False | Yes* |
--ancient_kmers_set |
Use ancient kmers text file instead of Bloom filter | False | Yes* |
--input_file |
Path to input FASTQ/FASTA file | None | Yes |
--output_prefix |
Prefix for output files (enables batch processing) | None | Yes |
--output |
Output directory path | output |
No |
--kmer_size |
K-mer size | 31 | No |
--n_consec_matches |
Number of consecutive matches to classify as anchor read | 2 | No |
--anchor_proportion_cutoff |
Minimum proportion of anchor k-mers to classify as ancient | 0.5 | No |
--ancient_bloom_capacity |
Capacity for Bloom filter if building from scratch | 2,000,000,000 | No |
*Either --ancient_bloom OR --ancient_kmers_set must be specified (mutually exclusive).
aKmerBroom requires:
Required:
- A FASTQ/FASTA file containing reads to be classified
- Either:
ancient_kmers.bloom: Pre-built Bloom filter with ancient k-mers (recommended)ancient_kmers: Text file with one ancient k-mer per line
The Bloom filter:
- The provided
data/ancient_kmers.bloomcontains trusted oral k-mers - Size: ~3.6GB, optimized for ancient oral DNA samples only
- Built from clean k-mers exclusive to modern and ancient oral samples
Input formats supported:
- FASTQ files (most common)
- FASTA files
aKmerBroom generates the following output files:
Main outputs:
{prefix}_annotated_reads.fastq: Intermediate output with anchor reads{prefix}_annotated_reads_with_anchor_kmers.fastq: Final classified readsaKmerBroom_{prefix}.log: Detailed processing log with statistics
Output file format: The final output FASTQ file contains reads with enhanced headers:
@SeqId ReadLen={length} ConsecMatch={true/false} AnchorProp={0.0-1.0}
Classification criteria:
- Reads with
AnchorProp≥ 0.5 (50%) are classified as ancient oral DNA - The threshold can be adjusted using
--anchor_proportion_cutoff
The tests/ folder contains a test dataset consisting of ancient oral data @SRR13355797 mixed with non-oral data @ERR671934.
Since the Bloom filter is now included in the repository, you can run a quick test:
# Activate environment
mamba activate aKmerBroom
# Quick test with included data and Bloom filter
aKmerBroom --ancient_bloom --input_file tests/unknown_reads.fastq --output_prefix test_sampleStep 1: Set up environment
# Create and activate the aKmerBroom environment
mamba env create -f environment.yml
mamba activate aKmerBroom
# Install as command-line tool
pip install -e .Step 2: Run test
# Test with the new command-line tool
aKmerBroom --ancient_bloom --input_file tests/unknown_reads.fastq --output_prefix test_runStep 3: Check results The results will be written to:
output/test_run_annotated_reads.fastq(intermediate output)output/test_run_annotated_reads_with_anchor_kmers.fastq(final output)aKmerBroom_test_run.log(log file with detailed statistics)