Latest:
Author of the study:
- Sara Bohnstedt Moerup (sara.bohnstedt.moerup@regionh.dk)
Initial Code Development by:
- Cameron MacPherson (cameron@biostone.consulting)
Modification of code and dockerisation by:
- Preston Leung (preston.yui.sum.leung@regionh.dk)
Additional contacts:
- Daniel Murray (daniel.dawson.murray@regionh.dk)
- Joanne Reekie (joanne.reekie@regionh.dk)
Please cite this paper when using this tool (To be updated).
Originally developed for testing SNPs within a single gene and its association with a specific outcome (used in this study by Murray, DD. et al1), this tool has been modified to include SNPs from different genes, allowing gene sets to be tested. Genes SKATO Analysis are a set of RScripts put into a docker such that it can be downloaded and executed on the user's computer without the need to install required packages.
You can follow the below instructions to obtain the copy of the working tool. However, if for some reason you cannot get a copy, please contact preston.yui.sum.leung@regionh.dk or daniel.dawson.murray@regionh.dk and we should be able to provide a .tar.gz version of the docker image.
The image that runs the analysis can be retrieved via:
docker pull pdawgzgg/gene_analysis_skato:[TAG]
where [TAG] is the version you would like to pull. (e.g. v:1.0)
docker run \
--rm \
-v /TRUE_PATH/TO/DATA:/dockerWorkDir/TORUN \
pdawgzgg/gene_analysis_skato:v1.0 \
-p /dockerWorkDir/TORUN/Analysis.parameters.csv \
-o /dockerWorkDir/TORUN/OUTPUT.csv
Note: The memory requirement could be quite intensive when dealing with large SNP files. Could potentially use upward of 36GB of memory.
| Docker Parameters | Description |
|---|---|
| --rm | Automatically remove the container when it exits |
| -v | Bind mount a volume |
| Required Parameters | Description |
|---|---|
| -o | Filename to write the output to. |
| -p | Filename of the input file. |
Note: In the above example command, the data required to run the tool is assumed be in /dockerWorkDir/TORUN/ folder or whatever name used to in place of TORUN. The output can be accessed in /TRUE_PATH/TO/DATA when it is completed.
In the input file used for the -p parameter, the .csv (comma-separated) is expected to store a dataframe columns about the data being used and the location of data.
An example of the parameter file would look like this:
| entrez | window | pidfile | confounder.1 | confounder.2 | confounder.3 | cohort | outcome | ignore | affy_file | pheno_file | geno_file |
|---|---|---|---|---|---|---|---|---|---|---|---|
| entrez ID | +/- window size from gene region | /PATH/TO/PIDFILE | Covariate 1 | Covariate 2 | Covariate 3 | COHORT-ID | Outcome Variable | 1 to ignore else 0 | /PATH/TO/AFFY | /PATH/TO/PHENO | /PATH/TO/GENO |
Note: If more covariates are to be needed for your dataset, simply add a column confounder.N where N is an integer.
| File Types | Description |
|---|---|
| pidfile | This is a list (.txt is fine) of subject/patient IDs that are to be included in the run. IDs are matched against IDs in pheno_file and geno_file. |
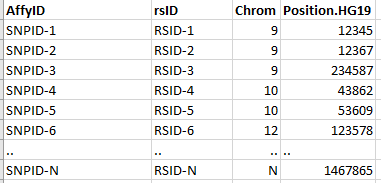
| affy_file | A dataframe-like file storing SNP information. Contains 4 columns: AffyID, rsID, chromosome and position stored in .RDS format. The AffyID column stores the SNP probe id name. This is the name that details the SNP count for each patient/subject. It may or may not be identical to rsID. |
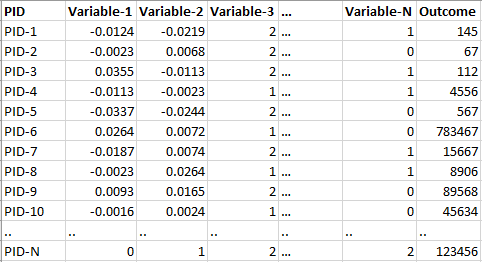
| pheno_file | A dataframe-like file storing the clinical data and/or phenotypes (columns) of each subject/patient (rows) stored in .RDS format. The confounder(s) and outcome names selected for analysis should be the same as the as column names within this file. |
| geno_file | A dataframe-like file storing subject/patient id (rows) by SNPs (columns). The number 1 or 2 represents the copies of SNP present and zero for absence for each subject/patient. Stored in .RDS format. The column names for the SNPs should be same as the AffyIDs in affy_file. |
geno_file and affy_file can be extracted from plink files using plink tool.
Briefly, geno_file is a modified form of the .raw file (this can be generated using plink tool). In the .raw file, IID is used distinguish subjects and is implemented as the rownames of the dataframe in the .RDS file. The columns FID, PAT, MAT, SEX and PHENOTYPE are not used. The following columns should be the SNP ids used by the SNP array (aka the labels used to identify Affymetrix SNP IDs). These Affymetrix SNP IDS will be reused in affy_file.
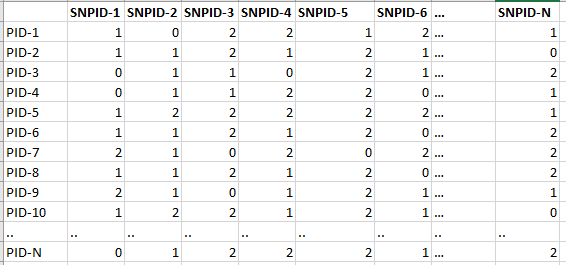
For affy_file, this can be extracted from the .bim file. The modification just adds an extra column for rsIDs, where the rsID is associated to the matching AffyID. AffyID and rsID does not need to be the same, hence the easiest way to link rsID to AffyID to use CHROM:POS:ALT:REF to match the two if you're using a SNP collection source like Ensembl GRCh37. Since AffyIDs are usually laboratory specific, linking it to rsID allows better access to information using standardised SNP identification.
Note: Please follow the column naming convention for affy_file.
For `pheno_file' it is quite straight forward. Please see below for an example of how it could look like:
For how to generate .RDS check out this blog.
R scripts (GeneAnalysis_SKATO.R, GeneAnalysis_SKATO_Helper.R and checkDependancies.R) are included in the repository if you wish to modify the code to suit your own analyses.
Additionally, the dockerfile (DockerScript/DockerFile) to build the image is also available for reference or modification if you wish to build your modified R scripts in to docker images for portability.
Archived Versions:
| Version | Repoistory | Notes |
|---|---|---|
| v1.0.0-PreRelease | Same base code. Incorrect tag. Recorded here for reference. | |
| v1.0-PreRelease | Current version. |
Please see R_PackageList.tsv in the repository for the full list of R packages used to build this tool.
I would like to give some special acknowledgement to Sara Bohnstedt Moerup for her effort and perseverance in learning and adapting to running analysis programs. As a clinician-scientist, it is not easy to jump into bioinformatics, especially when needing to deal with backend coding. Recognition of these efforts when there is a clear need to bridge the skill gap is often understated. So here, I wish express my appreciation and recognition of Sara's efforts to understand the messy side of bioinformatics in this project.
-Preston

This README file is written using Dillinger.
Footnotes
-
Murray, Daniel D.; Grund, Birgit,∗; MacPherson, Cameron R.,∗; Ekenberg, Christina; Zucco, Adrian G.; Reekie, Joanne; Dominguez-Dominguez, Lourdes; Leung, Preston; Fusco, Dahlene; Gras, Julien; Gerstoft, Jan; Helleberg, Marie; Borges, Álvaro H.; Polizzotto, Mark N.; Lundgren, Jens D. Association between ten-eleven methylcytosine dioxygenase 2 genetic variation and viral load in people with HIV. AIDS 37(3):p 379-387, March 1, 2023. | DOI: 10.1097/QAD.0000000000003427 ↩